
U.S. National Science Foundation Center for the Chemistry of Molecularly Optimized Networks (NSF MONET)

RECENT PUBLICATIONS
Follow NSF MONET's Research:
NSF MONET publications are both distinctive in scope and distinct from other funded research of Center members; all publications acknowledge NSF MONET and the U.S. National Science Foundations Centers for Chemical Innovation (CCI) program as the sole source of federal funding for MONET senior investigators.
Revealing the Topological Analogy between End-Linked and Pendant Cross-Linked Polymer Networks for Mechanophore-Enabled Toughening
K. Ko and J. A. Johnson
June 24, 2026

Recent advances in mechanochemistry have shown that the deliberate incorporation of mechanically weak bonds, specifically scissile mechanophores, can improve the fracture resistance of polymer networks. In pendant cross-linked elastomers (e.g., side-chain cross-linked or vulcanized elastomers), bifunctional mechanophores incorporated into cross-links produce substantial toughening. By contrast, utilizing the same motifs for toughening in end-linked or step-growth networks, which are ubiquitous in industrial thermosets, has remained a challenge. Here, by establishing a topological correspondence between pendant cross-linked and end-linked architectures, we identify a design strategy that enables bifunctional scissile motifs to successfully toughen end-linked networks. We validate this approach using model thermoset polyurethanes and show that the resulting mechanophore-driven toughening is substantial, yielding a 16-fold enhancement in tensile toughness and an 8-fold enhancement in tearing energy. We further show that this strategy can be combined with conventional polyurethane toughening approaches to produce materials with outstanding properties. More broadly, this work establishes strand continuity as a mechanochemical design principle for fracture resistant materials across diverse polymer architectures.
High-Throughput Discovery of Conformation-Switching Mechanophores with Enhanced Reactivity and Stability
X. Huang, R. St. Michel, I. Kevlishvili, and H. J. Kulik
June 6, 2026

Mechanophores offer unique opportunities in polymer chemistry, yet current mechanophores are often limited by low reactivity, irreversible transformation, or poor thermal stability. Here, we report the computational discovery of a new class of Cu2+ complex mechanophores comprising two tridentate scorpionate ligands that reversibly switch from octahedral to square-planar coordination under mechanical load. We curate over 750 synthetically accessible candidates using ligands from the Cambridge Structural Database. Leveraging density functional theory (DFT) and external force explicitly included (EFEI) modeling, we find 393 complexes that undergo conversion to square-planar coordination through concerted dissociation of two load-aligned coordination bonds at low applied force. We analyze the data to identify trends and characterize the most reactive mechanophores using interpretable machine learning (ML) models. This analysis reveals the most reactive mechanophores to be those that have chemical compositions leading to longer load-aligned and shorter load-orthogonal bonds as well as lower ligand steric bulk. Finally, we find robust candidates that exhibit both mechanical lability and thermal stability, with complexes comprising tris(2-pyridyl) ligands emerging as promising lead candidates. Together, this work establishes coordination switching as a useful design strategy for mechanophores and demonstrates how high-throughput ab initio screening and ML can enable data-driven discovery of force-responsive polymer building blocks.
Mechanophore cross-linking enhances ballistic energy dissipation of polymers
Z. Sang, S. T. Nguyen, K. Ko, S. Lin, H. Jang, S. Gonzalez-Zapata, S. Fitz, Y Kai, S. Kooi, C. Deng, M. Olvera de la Cruz, M. Koslowski, H. J. Kulik, S. L. Craig, K. A. Nelson, and J. A. Johnson
June 3, 2026

Mechanical failure is a marked limitation for plastics used in structural, protective and coating applications. In particular, perforation under high-rate deformation is difficult to mitigate through conventional molecular design1,2. Cross-linking is widely used to improve the thermal and chemical stability of polymers, yet under mechanical deformation, it typically renders materials more brittle, limiting impact resistance and functional lifetime3. Overcoming this fundamental trade-off between stability and toughness remains a central challenge. Here we demonstrate that embedding a small fraction of force-sensitive mechanophores as cross-links into common polymers fundamentally reverses this trade-off, producing materials with substantially enhanced ballistic energy dissipation. At strain rates exceeding 107 s−1, we show that mechanophore-cross-linked networks absorb up to about 115% more energy than conventional thermosets and surpass even their uncross-linked thermoplastic counterparts. We attribute this behaviour to a force- and adiabatic-heating-driven local thermoset-to-thermoplastic transition, in which selective mechanophore scission facilitates viscoplastic deformation at the impact site while preserving network integrity in the surrounding regions. We demonstrate the generality of this strategy in both glassy polystyrene and rubbery styrene–butadiene–styrene triblock copolymers. These results establish mechanophore cross-linking as a design principle for converting commodity polymers into impact-resilient materials and open directions at the intersection of polymer mechanochemistry and extreme-strain-rate material behaviour.
Role of Polymer–Protein Interactions in the Dynamics of Polymer-Integrated Protein Crystals
Y. Na, D. Le, P.-A. Lin, Z. Zhang, J. Pombo, F. Jimenez-Angeles, K. Han, L. Zhang, C. Do, H. T. Chiang, M. Olvera de la Cruz, A. Frano, F. Arya, and F. A. Tezcan
April 24, 2026

The incorporation of synthetic polymers into biomolecular materials provides a powerful strategy to enhance their properties. We recently showed that the interstitial spaces of highly solvated mesoporous ferritin crystals could be infiltrated with acrylate (Ac) and acrylamide (Am) monomers, which are subsequently polymerized in crystallo to yield a new class of hybrid materials termed Polymer-Integrated Protein Crystals (PIX). Our earlier studies had shown that ferritin-PIX displayed remarkable properties such as reversible expansion and contraction without losing crystalline order, efficient self-healing, and the ability to encapsulate and release large biomolecular cargo. However, the structure of the polyacrylate-co-acrylamide (p(Ac–Am)) polymer matrix, its distribution within the protein lattice, and the molecular nature of the protein–polymer interactions that ultimately engender the emergent properties of ferritin-PIX have remained unknown. Here, we combine small-angle neutron and X-ray scattering and analytical measurements with extensive all-atom and coarse-grained molecular dynamics simulations to examine the structure and dynamics of the polymer network within the crystalline framework of ferritin-PIX. Our results reveal an extensive and multivariate set of noncovalent interactions between the ferritin surfaces and p(Ac–Am) chains that sustain the structural coherence of the crystalline lattice while accommodating large-scale motions. Guided by these insights, we have demonstrated that changes in the chemical compositions of ferritin and the polymer matrix can be used to predictably control the structural dynamics of ferritin-PIX. Our increased molecular-level understanding and engineering of the polymer–protein interface in ferritin-PIX provide an important step toward the generalization of the PIX concept to other protein crystals and polymer compositions.
The Effect of Excluded Volume on Fracture Mechanisms in Coarse-Grained Simulations of Polymer Networks
D. Sen, K. Hasegawa, and B. D. Olsen
March 30, 2026

Macroscopic fracture behavior of polymer networks is governed by chain scission at the molecular level, yet the mechanism of fracture remains elusive. While state-of-the-art, highly coarse-grained simulation methods have been successful at predicting macroscopic fracture properties for strain rates of approximately 1/s, they are usually limited by key assumptions of periodic boundary conditions and purely attractive interactions within network elements. Incompressibility is enforced by constraining a constant lateral contraction of periodic boundary conditions to achieve Poisson’s ratio of 0.5 throughout the linear and nonlinear tensile regimes, which restricts the network structure and dynamics, preventing accurate analysis of fracture propagation at the molecular level. Here, a new coarse-grained fracture simulation approach is presented, which models repulsive interactions within a network as a mean-field excluded volume (EV) potential and eliminates the need for imposed deformation along nontensile axes. Simulation results reveal that consideration of EV interactions in networks leads to physically consistent and more realistic fracture behavior. Networks with EV exhibit a necking regime during fracture, consistent with the behavior of real networks under pure tensile deformation. Linear elastic moduli in networks with EV show excellent quantitative agreement with previously reported experimental results. While simulations without EV can predict correct quantitative trends in toughness, which is also reproduced in the simulations with EV, the molecular-level chain scission mechanisms obtained with EV are much more physically consistent, in stark qualitative and quantitative contrast to simulations without EV. Most notably, the presence of EV interactions facilitates accurate simulation of elastic recoil of the network after fracture, whereas the lack of EV leads to network collapse post-failure. EV interactions contribute to network homogenization during stress relaxation, leading to delayed fracture under low strains, whereas stress concentration is observed without EV.
Extending BigSMILES to Include Topological Bonds
H. R. Sudhakar, S. Craig, and B. D. Olsen
March 4, 2026

Machine-readable line notations such as SMILES are rapidly gaining popularity as a means of storing, searching, and analyzing chemical information. Within the SMILES framework, BigSMILES was developed to represent the stochastic connectivity characteristics of polymers. More recently, noncovalent BigSMILES extended the notation to encode noncovalent interactions, recognizing the stochastic nature of these bonds. Inspired by this framework, Topological BigSMILES is introduced to provide a further extension to represent topological interactions in macromolecules. This notation appends optional topological bond descriptors and associated indices, enabling the annotation of complex molecular architectures. In particular, the notation presented herein can encode the topological interactions found in knotted macrocycles and polymers, polycatenanes, and polyrotaxanes. The progression from BigSMILES to Topological BigSMILES highlights the potential for this framework to be used in representing broader classes of soft materials systems.
Optimizing the Stability of Viral Nanoparticles: Engineering Strategies, Applications, and the Emerging Concept of the Virophore
Z. Wu, J. L. Bayon, C. J. Ogilvie, S. L. Craig, and N. F. Steinmetz
January 8, 2026

Nanoparticles derived from plant viruses and bacteriophages are self-assembling structures that can be functionalized for broad applications in drug delivery, vaccine formulation, and imaging, as well as the engineering of nanomaterials, and nanoscale templating. Their capacity for precise chemical and genetic modification makes them versatile, but their functional potential may be limited by insufficient or poorly controlled structural stability. In this perspectives article, we assess recent advances in stability optimization to improve the functionality of virus-based nanoparticles and derived materials, considering engineering strategies that target the external and internal surfaces, as well as the interfaces between coat protein subunits. We look at approaches such as site-specific bioconjugation, reversible and permanent cross-linking, polymer endoskeleton reinforcement, metal ion coordination, and protective core–shell architectures, which can be used to tailor particle stability for harsh biological environments, or create particles with responsive stability profiles allowing smart delivery systems to release cargo when exposed to triggers such as pH, redox potential, illumination, or mechanical stress. We propose the concept of the virophore: a genetically or chemically encoded functional unit integrated into the structure of a virus particle that acts as a programmable structural switch, enabling reversible, triggerable reconfiguration in response to defined stimuli. We argue that embracing virophore design will expand the capabilities of virus-based nanomaterials beyond passive durability, facilitating adaptive, intelligent behavior. This will support applications such as programmable drug release, biosensors, and dynamic material systems.
Reimagining Polymer Networks from Molecule to Material
N. Nechmad, L. Campos, C. Deng, J. Diodati, S. Ekim, E. Garcia-Villatoro, J. P. Gong, A. Herzog-Rbeitman, X. Huang, J. Johnson, J. Kalow, R. Kemmerling, I. Kevlishvili, A. P. Kitos Vasconcelos, R. Klausen, H. J. Kulik, J. Leganes Bayon, M. Ma, E. McFee, J. Mendenhall, J. Moore, A. Nelson, J. Moore, B. Olsen, M. Rubinstein, C. Schindler, N. Sottos, N. Steinmetz, S. Wei, F. Xie, and S. L. Craig
December 12, 2025
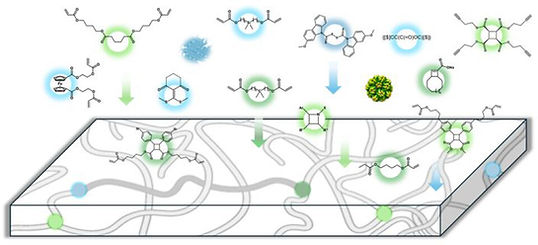
Polymer networks are complex materials with a broad distribution of molecular states, making it challenging to establish direct connections between the structure of individual network components and macroscopic material behavior. Recent advancements in mechanochemistry, dynamic covalent chemistry, and single-molecule characterization, however, are transforming this perspective. By leveraging the chemical reactivity of network junctions and integrating mechanophores into polymer networks, direct, quantitative links between molecular behavior and material properties are now being established. These insights are challenging long-standing property trade-offs and creating avenues for polymers with enhanced mechanical properties. This Perspective highlights how new molecule-to-material relationships in polymer networks are simultaneously advancing the field of polymer chemistry and uncovering fundamental principles that extend beyond polymer systems, enriching our broader understanding of reactivity, structure, and bonding in complex materials.
Tetrafunctional Cyclobutanes Tune Toughness via Network Strand Continuity
A. Herzog-Arbeitman, I. Kevlishvili, D. Sen, J. Lian, J. Chakraverty, S. Wang, B. D. Olsen, H. J. Kulik, S. L. Craig, and J. A. Johnson
November 14, 2025

Customizing the toughness of polymer networks independently of their chemical composition and topology remains an unsolved challenge. Traditionally, polymer network toughening is achieved by using specialized monomers or solvents or adding secondary networks/fillers that substantially alter the composition and may limit applications. Here we report a class of force-responsive molecules—tetrafunctional cyclobutanes (TCBs)—that enable the synthesis of single-network end-linked gels with substantially decreased or increased toughness, including unusually high toughness for dilute end-linked gels, with no other changes to network composition. This behaviour arises from stress-selective force-coupled TCB reactivity when stress is imparted from multiple directions simultaneously, which traditional bifunctional mechanophores cannot access. This molecular-scale mechanoreactivity translates to bulk toughness through a topological descriptor, network strand continuity, that describes the effect of TCB reactivity on the consequent local network topology. TCB mechanophores and the corresponding concepts of stress-selective force-coupled reactivity and strand continuity offer design principles for tuning the toughness of simple yet commonly used single-network gels.
Toughening 3D Printed Elastomers Using Mechanophore Crosslinkers
A. P. K. Vasconcelos, N. Van Zee, S. L. Craig, and A. Nelson
October 27, 2025

Elastomeric materials are widely used in industrial application sectors including construction, automotives, soft robotics, and biomedicine. Light-based three-dimensional (3D) printing enables the manufacturing of elastomeric polymer networks with geometric and functional customizability beyond the capabilities of traditional manufacturing methods. These 3D printed polymer networks often suffer from premature mechanical failure of the material that limits their viability in load-bearing applications. One approach to toughen elastomers is to employ non-covalent additives as sacrificial bonds in the polymer network; however, this toughness enhancement comes with a trade-off in the stiffness of the resultant object. Herein, we use a 1 : 1 substitution of cyclobutane-based mechanophores as scissile covalent crosslinks in 3D printed poly(methoxyethylacrylate) networks to enhance the material toughness without compromising stiffness. These crosslinkers increased the material's toughness in tensile and tearing tests without altering its stiffness or appearance. The enhanced toughness and tear resistance of these elastomers enabled bonding operations such as stitching and suturing. The results suggest that mechanophores offer a promising route to toughen 3D printed elastomers.
A Systematic Workflow for Mechanophore Design
F. Xie, Y. Sun, and J. S. Moore
September 25, 2025

Advancing mechanoresponsive materials require novel mechanophores, though clear and structured design guidelines are still emerging. In this work, we present a systematic workflow aimed at facilitating the design and discovery of new mechanophores. By integrating the classic iso-metrical CoGEF approach with our innovative iso-tensional Tension Model of Bond Activation (TMBA) simulation, the workflow described herein enables comprehensive evaluation of mechanophore candidates prior to experimental implementation, with a practical case study included for detailed illustration. This predictive capability allows computational screening, efficient identification and filtering away unexpected issues while providing valuable insights for potential structural optimization.
A Plant Virus Supramolecular NanoSponge for Delivery of Agricultural Pesticides to the Rhizosphere
Z. Wu, S. K. Hsu, N. F. Steinmetz
September 23, 2025

Managing parasitic plant pests is a significant burden on the global agricultural industry. Endoparasitic plant nematodes and phytopathogenic bacteria target and infect the plant rhizosphere, forming galls and reducing crop yields. Pesticide treatment is often inefficient because the pest-lethal pesticide dose must reach the root level, where pests infect the plants. We previously demonstrated that some plant viruses exhibit excellent soil mobility, and building on this, we here developed a viral NanoSponge formulation that enables the noncovalent loading of agrochemicals but prevents premature release through metal–phenolic network (MPN) coatings. Through optimized molecular interactions, the viral NanoSponge stabilized cargo binding and soil transport. Targeted cargo release and pest treatment were enabled through a pH-responsive cargo release mechanism. The NanoSponge demonstrated superior efficacy against nematodes (Caenorhabditis elegans) and against bacteria and biofilms thereof (Agrobacterium tumefaciens); efficacy was also demonstrated in bacteria-infected plants (Nicotiana benthamiana). The NanoSponge platform represents a sustainable and effective strategy for precision farming, effectively addressing deep soil pests.
CG-BigSMILES: Line Notation for Coarse-Grained Models of Polymers
B. S. Leao, W. Zou, N. J. Rebello, M. Rubinstein, L. F. M. Franco, and B. D. Olsen
August 27, 2025

The stochastic nature of polymers poses a challenge for developing machine-readable molecular representations, especially for coarse-grained computational models of materials. BigSMILES, a line notation that represents stochastic graphs, addresses this issue for atomistically resolved structures. Here, BigSMILES is extended to accommodate nonatomic particles, enabling it to represent coarse-grained models of polymers in a manner directly analogous to atomic structures. A new syntax, called coarse-grained BigSMILES (CG-BigSMILES), combines a layer-based annotation with linking to force field files to capture both chemical structures and interacting potentials, providing a complete description of coarse-grained models. The syntax allows optional definition of the mapping between the molecular structure and the coarse-grained “beads”, for both polymers and small molecules, in single- and multicomponent systems. CG-BigSMILES serves as a robust representation that allows researchers to unambiguously index coarse-grained systems in a manner analogous to that of atomistically resolved ones, enabling both types of systems to be integrated in single data structures and associated databases.
Design Principles of Stimuli-Responsive Covalent Adaptable Networks
Y. Xiong, C. Deng, S. Wei, L. M. Campos, and M. Olvera de la Cruz
August 20, 2025

Covalent adaptable networks (CANs) are sustainable polymeric materials that combine mechanical robustness with reprocessability through dynamic covalent bonds. Their mechanical behavior is governed by the kinetics and thermodynamics of bond reactions. Existing continuum models for CANs, based on classical rubber-like elasticity, often focus on steady-state behavior and overlook stress contributions from newly formed strands by assuming they are load-free. This limits their ability to predict behaviors like tensile recovery and actuation during transitions. To address these limitations, we develop a mean-field framework that connects microscopic bond reactions and molecular packing-induced conformational changes to macroscopic elastic behavior. Our model captures topological evolution and mechanical response of responsive networks undergoing bond reactions, validated using light-induced living polymer networks (LILPNs). To interpret the tensile recovery observed in LILPNs during transitions, we propose a conformation switch (CS) mechanism, which incorporates stress contributions from predeformed new strands due to molecular packing during transitions. The CS modeling reproduces key features of tensile recovery observed in experiments. Leveraging this framework, we also investigate the effects of molecular weight and functionality on LILPN dynamics. This work provides a predictive platform for designing responsive and adaptive polymer networks with tailored properties.
Tuning the Ultimate Strain of Single and Double Network Gels Through Reactive Strand Extension
X. Zheng, C.-Y. Chiou, S. D. Ekim, T. B. Kouznetsova, J. Vakil, Y. Hu, L. Sapir, D. Chen, Z. Wang, M. Rubinstein, J. P. Gong, N. R. Sottos, and S. L. Craig
August 15, 2025
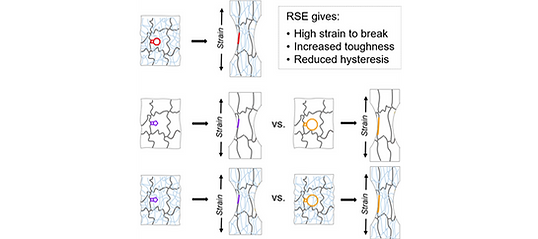
The stretchability (ability to be elongated) and toughness (capacity to absorb energy before breaking) of polymer network materials, such as elastomers and hydrogels, often determine their utility and lifetime. Direct correlations between the molecular behavior of polymer network components and the physical properties of the network inform the design of materials with enhanced performance, extended lifetime, and minimized waste stream. Here, we report the impact of the fused ring size in bicyclic cyclobutane mechanophores within the strands of polymer network gels. The mechanophores and their polymer strands share the same initial covalent contour length, whereas the capacity for reactive strand extension (RSE) is varied by changing the size of the ring fused to the cyclobutane from 5 to 12 carbon atoms. We observe the first evidence of covalent RSE effects in a single-network gel, and strands with greater RSE lead to gels with greater stretchability and toughness. The same qualitative correlation between molecular and macroscopic extension is also observed in DN hydrogels with mechanophores in the prestretched first network.
High-Throughput Discovery of Ferrocene Mechanophores with Enhanced Reactivity and Network Toughening
I. Kevlishvili, J. Vakil, D. W. Kastner, X. Huang, S. L. Craig, and H. J. Kulik
August 1, 2025

Mechanophores are molecules that undergo chemical changes in response to mechanical force, offering unique opportunities in chemistry, materials science, and drug delivery. However, many potential mechanophores remain unexplored. For example, ferrocenes are attractive targets as mechanophores due to their combination of high thermal stability and mechanochemical lability. However, the mechanochemical potential of ferrocene derivatives remains dramatically underexplored despite the synthesis of thousands of structurally diverse complexes. Herein, we report the computational, machine learning guided discovery of synthesizable ferrocene mechanophores. We identify over one hundred potential target ferrocene mechanophores with wide-ranging mechanochemical activity and use data-driven computational screening to identify a select number of promising complexes. We highlight design principles to alter their mechanochemical activation, including regio-controlled transition state stabilization through bulky groups and a change in mechanism through noncovalent ligand–ligand interactions. The computational screening is validated experimentally both at the polymer strand level through sonication experiments and at the network level, where a computationally discovered ferrocene mechanophore cross-linker leads to greater than 4-fold enhancement in material tearing energy. This work establishes a generalizable framework for the high-throughput discovery and rational design of mechanophores and offers insights into structure–activity relationships in mechanically responsive materials.
Rheological Isotope Effects for Molecular Insight in Covalent Adaptable
C. M. Hemmingsen, C. J. Chapman, C. Deng, Y. Xiong, C. J. Hanley, V. Zhang, M. Olvera de la Cruz, and J. A. Kalow
July 21, 2025

Understanding how small-molecule reactivity translates to bulk properties remains a fundamental challenge in polymer science. It is widely accepted that the viscoelastic behavior of covalent adaptable networks (CANs) is related to the kinetics of dynamic covalent bond exchange, but the nature of this relationship is complicated by the network environment. However, there are a lack of molecular tools to study exchange directly in polymer networks. In this work, we establish rheological isotope effects (RIEs) as a new tool for studying dynamic covalent exchange in bulk networks. By swelling dithioalkylidene-based CANs in H2O or D2O, we perturb the thiol/thiolate equilibrium to favor the reactive thiolate in D2O, accelerating exchange and thus stress relaxation rates. We found that bulk networks composed of thiol-terminated, multiarm poly(ethylene glycol) cross-linked by dithioalkylidenes exhibit a RIE between 0.31 and 0.64 for stress relaxation, depending on the cross-linker structure, cross-link density, and polymer topology. Monte Carlo simulations of network formation and evolution indicate that differences in RIE magnitude likely arise from topological defects including bridging chains. These defects reduce effective network size, decreasing the dependence of stress relaxation on bond exchange. We believe RIEs present new opportunities to study mechanisms of exchange in bulk networks, deconvolute modes of stress relaxation in complex CANs, and provide insight into the impact of defects on viscoelasticity.
Rubber that lasts longer
S. L. Craig and M. Rubinstein
May 7, 2025

Natural rubber has many uses in a variety of industries, enabled by ‘crosslinking’ between its tangled polymers, which creates elasticity. But rubber can crack and suffer fatigue. It is now shown that reducing the crosslink density in highly entangled natural rubber increases its crack resistance and prolongs its useful life.
Rapid self-strengthening in double-network hydrogels triggered by bond scission
Z. J. Wang, W. Li, X. Li, T. Nakajima, M. Rubinstein, and J. P. Gong
February 26, 2025

The scission of chemical bonds in materials can lead to catastrophic failure, with weak bonds typically undermining the materials’ strength. Here we demonstrate how weak bonds can be leveraged to achieve self-strengthening in polymer network materials. These weak sacrificial bonds trigger mechanochemical reactions, forming new networks rapidly enough to reinforce the material during deformation and significantly improve crack resistance. This rapid strengthening exhibits strong rate dependence, dictated by the interplay between bond breaking and the kinetics of force-induced network formation. As the network formation is generally applicable to diverse monomers and crosslinkers with different kinetics, a wide range of mechanical properties can be obtained. These findings may inspire the design of tough polymer materials with on-demand, rate-dependent mechanical behaviours through mechanochemistry, broadening their applications across various fields.
Enabling Selective Mechanochemical Scission of Network Crosslinks by Exchanging Single Carbon Atoms for Silicon
S. J. Melvin, Y. Yao, X. Huang, R. C. Bell, R. E. Kemmerling, I. Kevlishvili, A. C. Berg, A. P. Kitos Vasoncelos, A. Nelson, H. J. Kulik, S. L. Craig, and R. S. Klausen
February 4, 2025

The tearing of a polymer network arises from mechanochemically coupled bond-breaking events in the backbone of a polymer chain. An emerging research area is the identification of molecular strategies for network toughening, such as the strategic placement of mechanochemically reactive groups (e.g., scissile mechanophores) in the crosslinks of a network instead of in the load-bearing primary strands. These mechanically labile crosslinkers have typically relied on release of ring strain or weak covalent bonds for selective covalent bond scission. Here, we report a novel chemical design for accelerated mechanochemical bond scission based on replacing a single carbon atom in a crosslinker with a silicon atom. This single-atom replacement affords up to a two-fold increase in the tearing energy. We suggest a mechanism, validated by computational modeling, for accelerated mechanochemical Si–C bond scission based on minimizing the energy required to distort the starting material toward the transition-state geometry. We demonstrated the seamless incorporation of these scissile carbosilanes to toughen 3D-printed networks, which demonstrates their suitability for additive manufacturing processes.
Fracture of polymer-like networks with hybrid bond strengths
C. M. Hartquist, S. Wang, B. Deng, H. K. Beech, S. L. Craig, B. D. Olsen, M. Rubinstein, and X. Zhao
February 3, 2025

The design and functionality of polymeric materials hinge on failure resistance. While molecular-level details drive crack evolution in polymer networks, the connection between individual chain scission and bulk failure remains unclear and difficult to probe. In this work, we systematically study the fracture mechanics of polymer-like networks with hybrid bond strengths. We reveal that varying the ratio of strong and weak strands within otherwise identical networks gives a non-monotonic relationship between intrinsic fracture energy and strong strand fraction. Networks with some weak strands can counterintuitively outperform those with exclusively strong strands. Experiments on poly(ethylene glycol) gels and architected polymer-like lattices together with simulations unveil these properties. We show through computational visualization that strand type concentrations impact crack growth patterns and fracture energy trends. Cracks propagate through weak layers at low strong strand fractions. Aggregate clusters deflect or pin cracks at similar concentrations of strong and weak strands. Cracks blunt due to dispersed weak strand failure at high strong strand fractions. The sacrificial weak strands can notably deconcentrate stress near the crack tip, which toughens by delaying crack advancement. The interplay between concentration and clustering of strand types in networks with hybrid bond strengths, combined with crack growth phenomena and nonlocal energy release, provides insights into unusual fracture characteristics. Results shed light on fracture in polymer networks and percolated lattices.
Force-Activated Spin-Crossover in Fe2+ and Co2+ Transition Metal Mechanophores
X. Huang, I. Kevlishvili, S. L. Craig, and H. J. Kulik
December 23, 2024

ransition metal mechanophores exhibiting force-activated spin-crossover are attractive design targets, yet large-scale discovery of them has not been pursued due in large part to the time-consuming nature of trial-and-error experiments. Instead, we leverage density functional theory (DFT) and external force explicitly included (EFEI) modeling to study a set of 395 feasible Fe2+ and Co2+ mechanophore candidates with tridentate ligands that we curate from the Cambridge Structural Database. Among nitrogen-coordinating low-spin complexes, we observe the prevalence of spin crossover at moderate force, and we identify 155 Fe2+ and Co2+ spin-crossover mechanophores and derive their threshold force for low-spin to high-spin transition (FSCO). The calculations reveal strong correlations of FSCO with spin-splitting energies and coordination bond lengths, facilitating rapid prediction of FSCO using force-free DFT calculations. Then, among all Fe2+ and Co2+ spin-crossover mechanophores, we further identity 11 mechanophores that combine labile spin-crossover and good mechanical robustness that are thus predicted to be the most versatile for force-probing applications. We discover two classes of mer-symmetric complexes comprising specific heteroaromatic rings within extended π-conjugation that give rise to Fe2+ mechanophores with these characteristics. We expect the set of spin-crossover mechanophores, the design principles, and the computational approach to be useful in guiding the high-throughput discovery of transition metal mechanophores with diverse functionalities and broad applications, including mechanically activated catalysis.
Real-Time Quantification of Molecular-Level Dynamic Behaviors Underpinning Shear Thinning in End-Linked Associative Polymer Networks
Y. Zheng, D. Sen, W. Zou, K. Dai, and B. D. Olsen
December 11, 2024

Shear thinning of associative polymers is tied to bond breakage under deformation and retraction of dangling chains, as predicted by transient network theories. However, an in-depth understanding of the molecular mechanisms is limited by our ability to measure the molecular states of the polymers during deformation. Herein, utilizing a custom-built rheo-fluorescence setup, bond dissociation in model end-linked associative polymers is quantified in real time with nonlinear shear deformation based on a fluorescence quench transition when phenanthroline ligands bind with Ni2+. All of the networks exhibit shear thinning, and the dangling chain fraction increases with the shear rate. However, the number of broken bonds is smaller than that predicted by transient network theories, indicating additional relaxation modes or topological inhomogeneities in the networks. Through tuning counteranion chemistry, networks with similar relaxation times but varying dissociation and association rate constants (kd and ka) of Ni2+-phenanthroline cross-links are developed. Decreasing ka contributes to more dangling chain formation, while the effect of kd is less pronounced. Following force-accelerated bond dissociation of bridging chains, the dangling ends in networks with higher ka tend to reassociate to form elastically inactive loops, while the dangling chains are preserved in networks with lower ka. This indicates the critical role of bond reassociation kinetics in dictating shear-induced topological interchange of different chain configurations. Besides reaction kinetics, decreasing network junction functionality results in less shear thinning and broken bonds, originating from the lower amount of bond breakage required to flow and the higher tendency of the dissociated bonds to reform bridging chains.
Isomer-driven Polymerization, Depolymerization, and Reconstruction
H. Wakefield IV, N. J. Fromel, J. Jiang, I. Kevlishvili, Y. Yao, S. L. Craig, H. J. Kulik, and R. S. Klausen
November 20, 2024

We report that differences in ring strain enthalpy between cis and trans isomers of sila-cycloheptene provide a driving force for both polymerization and depolymerization via olefin metathesis. A need for new methods to reintroduce the low-strain isomer into the plastic economy inspired the development of a polymerization based on ring-opening/cross-metathesis step polymerization, which afforded perfect sequence control for an alternating copolymer. The chemical principles are a platform for achieving both efficient polymerization and depolymerization with high mass recovery in functional polymers.
The Restoring Force Triangle: A Mnemonic Device for Polymer Mechanochemistry
Y. Sun, F. Xie, and J. S. Moore
November 6, 2024

In polymer mechanochemistry, mechanophores are specific molecular units within the macromolecular backbone that are particularly sensitive to tension. To facilitate understanding of this selective responsiveness, we introduce the restoring force triangle (RFT). The RFT is a mnemonic device intended to provide intuitive insight into how external tensile forces (i.e., stretching) can selectively activate scissile bonds, thereby initiating mechanically driven chemical reactions. The RFT utilizes two easily computable parameters: the effective bond stiffness constant, which measures a bond’s resistance to elongation, and the bond dissociation energy, which is the energy required to break a bond. These parameters help categorize reactivity into thermal and mechanical domains, providing a useful framework for developing new mechanophores that are responsive to force but thermally stable. The RFT helps chemists intuitively understand how tensile force contributes to the activation of a putative mechanophore, facilitating the development of mechanochemical reactions and mechano-responsive materials.
Strain Learning in Protein-based Mechanical Metamaterials
N. Sadaba, E. Sanchez-Rexach, C. Waltmann, M. Olvera de la Cruz, L. Meza, H. Sardon, and A. Nelson
October 30, 2024

We established a strain learning mechanical metamaterial that can not only recover after plastic deformation but also become stronger and stiffer in response to the applied loads. We demonstrate that shape-memory protein–polymer networks can unravel to release their stored length and, after recovery, will reconfigure to accommodate higher loads. These materials are incorporated into lattice frameworks using additive manufacturing which are then shown to become up to 2.5× stiffer after being crushed to 80% strain and recovered. This strain learning behavior enables the creation of metamaterials that are “teachable” and can autonomously remodel in response to applied loads, similar to the processes that occur in natural materials.
Caged AIEgens: Multicolor and White Emission Triggered by Mechanical Activation
Y. Sun, K. Wang, X. Huang, S. Wei, E. Contreras, P. K. Jain, L. M. Campos, H. J. Kulik, and J. S. Moore
September 22, 2024

Aggregation-induced emission luminogens (AIEgens) that respond to mechanical force are increasingly used as force probes, memory devices, and advanced security systems. Most of the known mechanisms to modulate mechanoresponsive AIEgens have been based on changes in aggregation states, involving only physical alterations. Instances that employ covalent bond cleavage are still rare. We have developed a novel mechanochemical uncaging strategy to unveil AIEgens with diverse emission characteristics using engineered norborn-2-en-7-one (NEO) mechanophores. These NEO mechanophores were covalently integrated into polymer molecules and activated in both the solution and solid states. This activation resulted in highly tunable fluorescence upon immobilization through solidification or aggregation, producing blue, green, yellow, and orange-red emissions. By designing the caged and uncaged forms as donor–acceptor pairs for Förster resonance energy transfer (FRET), we achieved multicolor mechanofluorescence, effectively broadening the color spectrum to include white emission. Additionally, we computationally explored the electronic structures of activated NEOs, providing insights into the observed regiochemical effects of the substituents. This understanding, together with the novel luminogenic characteristics of the caged and activated species, provides a highly tunable reporter that traces progress with continuous color evolution. This advancement paves the way for future applications of mechanoresponsive materials in areas like damage detection and bioimaging.
Toughening and Imparting Deconstructability to 3D Printed Photopolymer Resins with “Transferinker” Additives
K. P. Qin, A. Herzog-Arbeitman, W. Zou, S. Chakraborty, S. L. Kristufek, K. E. L. Husted, S. L. Craig, G. D. Joly, B. D. Olsen, and J. A. Johnson
September 11, 2024

Thermoset toughness and deconstructability are often opposing features; simultaneously improving both without sacrificing other mechanical properties (e.g., stiffness and tensile strength) is difficult, but, if achieved, could enhance the usage lifetime and end-of-life options for these materials. Here, a strategy that addresses this challenge in the context of photopolymer resins commonly used for 3D printing of glassy, acrylic thermosets is introduced. It is shown that incorporating bis-acrylate “transferinkers,” which are cross-linkers capable of undergoing degenerative chain transfer and new strand growth, as additives (5–25 mol%) into homemade or commercially available photopolymer resins leads to photopolymer thermosets with substantially improved tensile toughness and triggered chemical deconstructability with minimal impacts on Young's moduli, tensile strengths, and glass transition temperatures. These properties result from a transferinker-driven topological transition in network structure from the densely cross-linked long, heterogeneous primary strands of traditional photopolymer networks to more uniform, star-like networks with few dangling ends; the latter structure more effectively bear stress yet is also more easily depercolated via solvolysis. Thus, transferinkers represent a simple and effective strategy for improving the mechanical properties of photopolymer thermosets and providing a mechanism for their triggered deconstructability.
Digital Light Processing (DLP) 3D Printing of Polymer Networks Comprising Virus-Like Particles
N. Sadaba, J. L. Bayón, A. Nelson, and N. F. Steinmetz
July 22, 2024

In this work, we introduce a 3D-printable virus-like particle (VLP)-enhanced cross-linked biopolymer system. VLPs displaying surface-available acrylate groups were prepared through aza-Michael addition to serve as resins. The VLP resins were then photopolymerized into a poly(ethylene glycol) diacrylate (PEGDA) network following DLP 3D printing. This approach represents a convergence of disciplines, where the synergistic interaction between virology and additive manufacturing unlocks new frontiers in biotechnology.
Visible-Light-Mediated aza Paternò-Büchi Reaction of Acyclic Oximes and Alkenes for the Synthesis of Monocyclic Azetidines
E. Wearing, Y.-C. Yeh, G. Terrones, S. Parikh, I. Kevlishvili, H. J. Kulik, and C. Schindler
June 27, 2024

The aza Paternò–Büchi reaction is a [2+2]-cycloaddition reaction between imines and alkenes that produces azetidines, four-membered nitrogen-containing heterocycles. Currently, successful examples rely primarily on either intramolecular variants or cyclic imine equivalents. To unlock the full synthetic potential of aza Paternò–Büchi reactions, it is essential to extend the reaction to acyclic imine equivalents. Here, we report that matching of the frontier molecular orbital energies of alkenes with those of acyclic oximes enables visible light–mediated aza Paternò–Büchi reactions through triplet energy transfer catalysis. The utility of this reaction is further showcased in the synthesis of epi-penaresidin B. Density functional theory computations reveal that a competition between the desired [2+2]-cycloaddition and alkene dimerization determines the success of the reaction. Frontier orbital energy matching between the reactive components lowers transition-state energy (ΔGǂ) values and ultimately promotes reactivity.
The Tension-Activated Carbon–Carbon Bond
Y. Sun, I. Kevlishvili, T. B. Kouznetsova, Z. P. Burke, H. J. Kulik, S. L. Craig, and J. S. Moore
June 13, 2024

Mechanical force drives distinct chemical reactions; yet, its vectoral nature results in complicated coupling with reaction trajectories. Here, we utilize a physical organic model inspired by the classical Morse potential and its differential forms to identify effective force constant (keff) and reaction energy (ΔE) as key molecular features that govern mechanochemical kinetics. Through a comprehensive experimental and computational investigation with four norborn-2-en-7-one (NEO) mechanophores, we establish the relationship between these features and the force-dependent energetic changes along the reaction pathways. We show that the complex kinetic behavior of the tensioned bonds is generally and quantitatively predicted by a simple multivariate linear regression based on the two easily computed features with a straightforward workflow. These results demonstrate a general mechanistic framework for mechanochemical reactions under tensile force and provide a highly accessible tool for the large-scale computational screening in the design of mechanophores.
A Simple Swell-and-Click Method for the Covalent Attachment of Virus-like Particles to Polymer Hydrogels
J. L. Bayon, C. Shih, S. L. Craig, and N. F. Steinmetz
June 1, 2024

Plant virus-like particles (VLPs) are biocompatible, non-infectious nanomaterials with promising applications as immunotherapeutics and vaccines. However, slow-release VLP formulations are needed to achieve long-term efficacy without repeated administration. VLP hydrogels allow the encapsulation and sustained delivery of VLPs, but the particles must covalently bind the hydrogel polymers to avoid premature loss. This has been achieved so far by in situ VLP polymerization, which requires high viral concentrations (5–10 mg/mL, 0.5–1 wt%) to form stable hybrid VLP–hydrogel networks and this complicates scalability and clinical translation. Here, we developed a novel swell-and-click method that led to successful VLP scaffold formation regardless of the viral load used. As a result, VLP-functionalized hydrogels were fabricated with viral concentrations as low as 0.1–1 mg/mL (0.01–0.1 % wt%) without compromising the scaffold stability on the process. The hydrogels incorporate VLPs during swelling, followed by copper-free click chemistry reactions that bind the particles covalently to the polymer. The swell-and-click method also resulted in more than a two-fold enhancement in VLP uptake into the hydrogels and it provides a means of combined burst release and prolonged sustained release, desired traits for cancer immunotherapy treatment. The present work introduces a novel methodology for the design of VLP-based hydrogels, which could facilitate the scalability of the fabrication process and move a significant step forward towards clinical translation of long-term VLP vaccination in cancer disease.
Valence Can Control the Nonexponential Viscoelastic Relaxation of Multivalent Reversible Gels
H. Le Roy, J. Song, D. Lundberg, A. V. Zhukhovitskiy, J. A. Johnson, G. H. McKinley, N. Holten-Andersen, and M. Lenz
May 15, 2024

Gels made of telechelic polymers connected by reversible cross-linkers are a versatile design platform for biocompatible viscoelastic materials. Their linear response to a step strain displays a fast, near-exponential relaxation when using low-valence cross-linkers, while larger supramolecular cross-linkers bring about much slower dynamics involving a wide distribution of timescales whose physical origin is still debated. Here, we propose a model where the relaxation of polymer gels in the dilute regime originates from elementary events in which the bonds connecting two neighboring cross-linkers all disconnect. Larger cross-linkers allow for a greater average number of bonds connecting them but also generate more heterogeneity. We characterize the resulting distribution of relaxation timescales analytically and accurately reproduce stress relaxation measurements on metal-coordinated hydrogels with a variety of cross-linker sizes including ions, metal-organic cages, and nanoparticles. Our approach is simple enough to be extended to any cross-linker size and could thus be harnessed for the rational design of complex viscoelastic materials.
Nested Non-Covalent Interactions Expand the Functions of Supramolecular Polymer Networks
D. Lundberg, C. Brown, E. Bobylev, N. Oldenhuis, Y. AlFaraj, J. Zhao, I. Kevlishvili, H. J. Kulik, and J. A. Johnson
May 10, 2024

Supramolecular polymer networks contain non-covalent cross-links that enable access to broadly tunable mechanical properties and stimuli-responsive behaviors; the incorporation of multiple unique non-covalent cross-links within such materials further expands their mechanical responses and functionality. To date, however, the design of such materials has been accomplished through discrete combinations of distinct interaction types in series, limiting materials design logic. Here we introduce the concept of leveraging “nested” supramolecular crosslinks, wherein two distinct types of non-covalent interactions exist in parallel, to control bulk material functions. To demonstrate this concept, we use polymer-linked Pd2L4 metal–organic cage (polyMOC) gels that form hollow metal–organic cage junctions through metal–ligand coordination and can exhibit well-defined host-guest binding within their cavity. In these “nested” supramolecular network junctions, the thermodynamics of host-guest interactions within the junctions affect the metal–ligand interactions that form those junctions, ultimately translating to substantial guest-dependent changes in bulk material properties that could not be achieved in traditional supramolecular networks with multiple interactions in series.
Internal Catalysis in Dynamic Hydrogels with Associative Thioester Cross-Links
V. Zhang, C. Ou, I. Kevlishvili, J. V. Accardo, H. J. Kulik, and J. A. Kalow
May 3, 2024

Thioesters are an essential functional group in biosynthetic pathways, which has motivated their development as reactive handles in probes and peptide assembly. Thioester exchange is typically accelerated by catalysts or elevated pH. Here, we report the use of bifunctional aromatic thioesters as dynamic covalent cross-links in hydrogels, demonstrating that at physiologic pH in aqueous conditions, transthioesterification facilitates stress relaxation on the time scale of hundreds of seconds. We show that intramolecular hydrogen bonding is responsible for accelerated exchange, evident in both molecular kinetics and macromolecular stress relaxation. Drawing from concepts in the vitrimer literature, this system exemplifies how dynamic cross-links that exchange through an associative mechanism enable tunable stress relaxation without altering stiffness.
Effect of the Activation Force of Mechanophore on Its Activation Selectivity and Efficiency in Polymer Networks
Z.J. Wang, S. Wang, J. Jiang, Y. Hu, T. Nakajima, S. Maeda, S. L. Craig, and J. P. Gong
May 2, 2024

In recent decades, more than 100 different mechanophores with a broad range of activation forces have been developed. For various applications of mechanophores in polymer materials, it is crucial to selectively activate the mechanophores with high efficiency, avoiding nonspecific bond scission of the material. In this study, we embedded cyclobutane-based mechanophore cross-linkers (I and II) with varied activation forces (fa) in the first network of the double network hydrogels and quantitively investigated the activation selectivity and efficiency of these mechanophores. Our findings revealed that cross-linker I, with a lower activation force relative to the bonds in the polymer main chain (fa-I/fa-chain = 0.8 nN/3.4 nN), achieved efficient activation with 100% selectivity. Conversely, an increase of the activation force of mechanophore II (fa-II/fa-chain = 2.5 nN/3.4 nN) led to a significant decrease of its activation efficiency, accompanied by a substantial number of nonspecific bond scission events. Furthermore, with the coexistence of two cross-linkers, significantly different activation forces resulted in the almost complete suppression of the higher-force one (i.e., I and III, fa-I/fa-III = 0.8 nN/3.4 nN), while similar activation forces led to simultaneous activations with moderate efficiencies (i.e., I and IV, fa-I/fa-IV = 0.8 nN/1.6 nN). These findings provide insights into the prevention of nonspecific bond rupture during mechanophore activation and enhance our understanding of the damage mechanism within polymer networks when using mechanophores as detectors. Besides, it establishes a principle for combining different mechanophores to design multiple mechanoresponsive functional materials.
Visible-Light-Induced Living Polymer Networks for Reconfiguration and Post-Transformation
S. Wei, J. Smith-Jones, R. F. Lalisse, J. Hestenes, E. Churchill, N. Munich, S. Danielsen, D. Chen, L. Marbella, O. Gutierrez, M. Rubinstein, A. Nelson, and L. M. Campos
April 9, 2024

The advent of covalent adaptable networks (CANs) through the incorporation of dynamic covalent bonds has led to unprecedented properties of macromolecular systems, which can be engineered at the molecular level. Among the various types of stimuli that can be used to trigger chemical changes within polymer networks, light stands out for its remote and spatiotemporal control under ambient conditions. However, most examples of photoactive CANs need to be transparent and they exhibit slow response, side reactions, and limited light penetration. In this vein, it is interesting to understand how molecular engineering of optically active dynamic linkages that offer fast response to visible light can impart “living” characteristics to CANs, especially in opaque systems. Here, the use of carbazole-based thiuram disulfides (CTDs) that offer dual reactivity as photoactivated reshuffling linkages and iniferters under visible light irradiation is reported. The fast response to visible light activation of the CTDs leads to temporal control of shape manipulation, healing, and chain extension in the polymer networks, despite the lack of optical transparency. This strategy charts a promising avenue for manipulating multifunctional photoactivated CANs in a controlled manner.
A Thermally Stable SO2 Releasing Mechanophore: Facile Activation, Single-Event Spectroscopy, and Molecular Dynamic Simulations
Y. Sun, W. J. Neary, X. Huang, T. B. Kouznetsova, T. Ouchi, I. Kevlishvili, K. Wang, Y. Chen, H. J. Kulik, S. L. Craig, and J. S. Moore
April 6, 2024

Polymers that release small molecules in response to mechanical force are promising candidates as next-generation on-demand delivery systems. Despite advancements in the development of mechanophores for releasing diverse payloads through careful molecular design, the availability of scaffolds capable of discharging biomedically significant cargos in substantial quantities remains scarce. In this report, we detail a nonscissile mechanophore built from an 8-thiabicyclo[3.2.1]octane 8,8-dioxide (TBO) motif that releases one equivalent of sulfur dioxide (SO2) from each repeat unit. The TBO mechanophore exhibits high thermal stability but is activated mechanochemically using solution ultrasonication in either organic solvent or aqueous media with up to 63% efficiency, equating to 206 molecules of SO2 released per 143.3 kDa chain. We quantified the mechanochemical reactivity of TBO by single-molecule force spectroscopy and resolved its single-event activation. The force-coupled rate constant for TBO opening reaches ∼9.0 s–1 at ∼1520 pN, and each reaction of a single TBO domain releases a stored length of ∼0.68 nm. We investigated the mechanism of TBO activation using ab initio steered molecular dynamic simulations and rationalized the observed stereoselectivity. These comprehensive studies of the TBO mechanophore provide a mechanically coupled mechanism of multi-SO2 release from one polymer chain, facilitating the translation of polymer mechanochemistry to potential biomedical applications.
Angle-Strained Sila-Cycloalkynes
H. Wakefield, S. J. Melvin, J. Jiang, I. Kevlishvili, M. A. Siegler, S. L. Craig, H. J. Kulik, and R. S. Klausen
April 5, 2024

Second row elements in small- and medium-rings modulate strain. Herein we report the synthesis of two novel oligosilyl-containing cycloalkynes that exhibit angle-strain, as observed by X-ray crystallography. However, the angle-strained sila-cyclooctynes are sluggish participants in cycloadditions with benzyl azide. A distortion-interaction model analysis based on density functional theory calculations was performed.
Self-Amplified HF Release and Polymer Deconstruction Cascades Triggered by Mechanical Force
Y. Hu, L. Wang, I. Kevlishvili, S. Wang, C.-Y. Chious, P. Shieh, Y. Ling, H. J. Kulik, J. A. Johnson, and S. L. Craig
March 30, 2024

Hydrogen fluoride (HF) is a versatile reagent for material transformation, with applications in self-immolative polymers, remodeled siloxanes, and degradable polymers. The responsive in situ generation of HF in materials therefore holds promise for new classes of adaptive material systems. Here, we report the mechanochemically coupled generation of HF from alkoxy-gem-difluorocyclopropane (gDFC) mechanophores derived from the addition of difluorocarbene to enol ethers. Production of HF involves an initial mechanochemically assisted rearrangement of gDFC mechanophore to α-fluoro allyl ether whose regiochemistry involves preferential migration of fluoride to the alkoxy-substituted carbon, and ab initio steered molecular dynamics simulations reproduce the observed selectivity and offer insights into the mechanism. When the alkoxy gDFC mechanophore is derived from poly(dihydrofuran), the α-fluoro allyl ether undergoes subsequent hydrolysis to generate 1 equiv of HF and cleave the polymer chain. The hydrolysis is accelerated via acid catalysis, leading to self-amplifying HF generation and concomitant polymer degradation. The mechanically generated HF can be used in combination with fluoride indicators to generate an optical response and to degrade polybutadiene with embedded HF-cleavable silyl ethers (11 mol %). The alkoxy-gDFC mechanophore thus provides a mechanically coupled mechanism of releasing HF for polymer remodeling pathways that complements previous thermally driven mechanisms.
Visible Light-Mediated (2+2)-Cycloadditions for the Formation of Macrocyclic Dimers
C. Ng, S. Kim, I. Kevlishvili, S. Terrones, E. Wearing, H. Kulik, and C. Schindler
March 4, 2024

Macrocyclic dimeric lactones have pharmacological activities that make them attractive synthetic targets, but typical synthetic strategies employ an iterative approach to construct the macrocycle. Herein, we report a visible-light-mediated approach that enables facile access to 1- and 2-azetine-based dimeric lactones of up to 30-membered ring macrocycles. These products form via four consecutive triplet energy transfers for 1-azetine dimeric products and two consecutive triplet energy transfers for 2-azetine dimeric products. Computational investigations provide insights into the mechanism of this reaction, consistent with an unexpected initial intermolecular [2 + 2]-cycloaddition being preferred under nonstandard Curtin–Hammett conditions over the corresponding intramolecular reaction, which ultimately enables an efficient reaction pathway for macrocyclic dimerization.
3D Printed Modular Piezoionic Sensors Using Dynamic Covalent Bonds
J. Smith-Jones, N. Ballinger, N. Sadaba, X. Lopez de Pariza, Y. Yao, S. L. Craig, H. Sardon, and A. Nelson
February 20, 2024

Flexible and lightweight sensors can assess their environment for a broad range of applications that include wearables for health monitoring and soft robotics. While 2D and 3D printing enables control over sensor design in multiple dimensions, customizability of a sensor toward different individual use cases is still limited because each sensor requires a new design and manufacturing process. Thus, there is a need for methodologies that produce modular sensor components that can be assembled and customized by an individual user. Herein, we demonstrate 3D printed, elastomeric ionogels comprising covalent adaptable networks (CANs) for modular sensor assemblies. Reversible Diels–Alder connections incorporated into the network can occur at the interface between two 3D printed objects in physical contact with each other. As a result, modular components can be combined and assembled on-demand into customized piezoionic sensors. Thermal curing of these modular blocks triggered the dynamic remodeling of the polymer networks that caused them to become fused together. Three different configurations (linear, cyclic, and box assemblies) were demonstrated to afford piezoionic sensors from the same set of 3D printed building blocks. This study highlights the benefits of dynamic covalent networks toward decentralized manufacturing, wherein a modular approach enables customization of 3D printed parts without the need for modifying the original design.
Oligosiloxane-Based Epoxy Vitrimers: Adaptable Thermosetting Networks with Dual Dynamic Bonds
A. A. Putnam-Neeb, A. Stafford, S. Babu, S. J. Chapman, C. M. Hemmingsen, M. S. Islam, A. K. Roy, J. A. Kalow, V. Varshney, D. Nepal, and L. A. Baldwin
February 7, 2024

Embedding dynamic covalent bonds into polymer compositions transforms static thermosets into active materials with the reprocessability of thermoplastics and the bulk properties of cross-linked networks. This class of next-generation materials, called covalent adaptable networks, shows significant promise in composites, soft optoelectronics, and robotics. Herein, we synthesized two oligosiloxane-based epoxy networks that provide fast dynamic bond exchange. Oligosiloxane diepoxides were cured with stoichiometric amounts of 1,2-phenylenediacetic acid to generate epoxy acid networks with two dynamic covalent bonding mechanisms. The resulting polymer networks provided access to fast stress-relaxation times (1–10 min) at temperatures of only 130 °C with excellent reprocessability.
Virus-like Particles Armored by an Endoskeleton
Z. Wu, J. L. Bayón, T. B. Kouznetsova, T. Ouchi, K. J. Barkovich, S. K. Hsu, S. L. Craig, and N. F. Steinmetz
January 31, 2024

Many virus-like particles (VLPs) have good chemical, thermal, and mechanical stabilities compared to those of other biologics. However, their stability needs to be improved for the commercialization and use in translation of VLP-based materials. We developed an endoskeleton-armored strategy for enhancing VLP stability. Specifically, the VLPs of physalis mottle virus (PhMV) and Qβ were used to demonstrate this concept. We built an internal polymer “backbone” using a maleimide–PEG15–maleimide cross-linker to covalently interlink viral coat proteins inside the capsid cavity, while the native VLPs are held together by only noncovalent bonding between subunits. Endoskeleton-armored VLPs exhibited significantly improved thermal stability (95 °C for 15 min), increased resistance to denaturants (i.e., surfactants, pHs, chemical denaturants, and organic solvents), and enhanced mechanical performance. Single-molecule force spectroscopy demonstrated a 6-fold increase in rupture distance and a 1.9-fold increase in rupture force of endoskeleton-armored PhMV. Overall, this endoskeleton-armored strategy provides more opportunities for the development and applications of materials.
Roadmap for Soft Matter: Stimuli Responsive Materials
A. Nelson and S. L. Craig
December 12, 2023

The term 'stimuli-responsive' (SR) refers to materials that undergo a meaningful change in properties (the response) when subjected to a change in external environment (the stimulus). The term is intrinsically redundant—a response by definition has a stimulus that triggered it, and the delivery of energy or matter is only a stimulus if it triggers a response. The broad, if linguistically uneconomical, use of 'stimuli-responsive' likely reflects a desire to emphasise the breadth available on either side of the stimulus–response relationship. Stimuli include various forms of energy (e.g. thermal, photons, electromagnetic waves and fields, or mechanical) and the introduction or removal of matter (e.g. solvents, reagents, acids/bases, salt, or electrons), and each can in principle be coupled to an ever-increasing range of responses (e.g. change in shape, assembly, colour, luminescence, mechanical properties, thermal transport, material transport, conduction, or uptake/release of cargo). The interest in, and study of, such materials has captured commercial and academic interest for decades. Excellent reviews of stimuli-responsive materials are available.
Reactivity-Guided Depercolation Processes Determine Fracture Behavior in End-Linked Polymer Networks
H. K. Beech, S. Wang, D. Sen, D. Rota, T. B. Kouznetsova, A. Arora, M. Rubinstein, S. L. Craig, and B. D. Olsen
December 1, 2023

The fracture of polymer networks is tied to the molecular behavior of strands within the network, yet the specific molecular-level processes that determine the mechanical limits of a network remain elusive. Here, the question of reactivity-guided fracture is explored in otherwise indistinguishable end-linked networks by tuning the relative composition of strands with two different mechanochemical reactivities. Increasing the substitution of less mechanochemically reactive (“strong”) strands into a network comprising more reactive (“weak”) strands has a negligible impact on the fracture energy until the strong strand content reaches approximately 45%, at which point the fracture energy sharply increases with strong strand content. This aligns with the measured strong strand percolation threshold of 48 ± 3%, revealing that depercolation, or the loss of a percolated network structure, is a necessary criterion for crack propagation in a polymer network. Coarse-grained fracture simulations agree closely with the tearing energy trend observed experimentally, confirming that weak strand scissions dominate the failure until the strong strands approach percolation. The simulations further show that twice as many strands break in a mixture than in a pure network.
Kinetic Model for Off-Stoichiometric Cross-Linking Reactions of End-Linked Polymer Networks
H. K. Beech, T.-S. Lin, H. Mochigase, and B. D. Olsen
November 27, 2023

The formation of end-linked polymer networks is commonly modeled as idealized chemical reactions, resulting in defect-free networks. However, many widely used industrial processes including platinum-catalyzed vinyl-silane cross-linking of poly(dimethylsiloxane) (PDMS) are mechanistically complex and involve a variety of side reactions. Here, a kinetic graph theory (KGT) model was updated to account for off-stoichiometric reactive groups and side reactions by adding two fitting parameters representing the relative rate of competing side reactions and the probability of side cross-linking events. The updated KGT outputs the population of each junction type from which the reaction fates of both starting materials are calculated. The elastic effectiveness of the resulting network is calculated with the nonlinear Miller–Macosko theory (MMT), updated to account for side reactions and side cross-linking. The MMT was validated on off-stoichiometric data and was chosen here for its ability to account for a range of effective junction functionalities. Combined, the updated KGT and MMT provide elasticity estimates that capture the experimental peak in elastic modulus observed at an off-stoichiometric silane/alkene ratio in PDMS networks. Both the Lake Thomas and micronetwork fracture theories were subsequently used to estimate the tearing energy, showing a similar peak at off-stoichiometric ratios in qualitative agreement with experimental data. This model is useful in systems where the cross-linking chemistry yields more complex reaction networks, making it relevant to many classes of polymer network chemistry where classical theories may not adequately capture network behavior.
Kinetics of Polymer Gel Formation Cause Deviation from Percolation Theory in the Dilute Regime
H. K. Beech, T.-S. Lin, D. Sen, D. Rota, and B. D. Olsen
November 14, 2023

Gelation has long been conceptualized and modeled as a percolation process in which bond formation or destruction events are random. Percolation assumes that connections are created or destroyed randomly such that the critical point should occur at the same point when approached from either direction. Here, the gel point of an end-linked poly(ethylene glycol) gel was measured during forward (bond forming) and reverse (bond breaking) gelation and degelation processes to interrogate how the gel point scales with synthesis concentration, where decreased concentration leads to an increased prevalence of inelastic loops. Forward gel points, measured with combined kinetic nuclear magnetic resonance (NMR) and diffusing wave spectroscopy (DWS) experiments, were identical to results generated from a kinetic Monte Carlo (KMC) simulation, demonstrating the expected gel point suppression as the concentration decreased. Reverse gel points, measured with a selective degradation technique, were within the error of forward gel points at high concentrations but displayed a lesser degree of suppression as the concentration decreased. This deviation between forward and reverse gel points at low concentrations was qualitatively reproduced in the KMC simulation. These experiments and simulations show that forward and reverse gel points diverge as the gel system becomes more dilute, suggesting that kinetic effects cause a departure from the percolation behavior in defect-rich gels.
Thermally Robust yet Deconstructable and Chemically Recyclable High-Density Polyethylene (HDPE)-Like Materials Based on Si−O Bonds
A. Johnson and J. Johnson
October 30, 2023

Polyethylene (PE) is the most widely produced synthetic polymer. By installing chemically cleavable bonds into the backbone of PE, it is possible to produce chemically deconstructable PE derivatives; to date, however, such designs have primarily relied on carbonyl- and olefin-related functional groups. Bifunctional silyl ethers (BSEs; SiR2(OR′2)) could expand the functional scope of PE mimics as they possess strong Si−O bonds and facile chemical tunability. Here, we report BSE-containing high-density polyethylene (HDPE)-like materials synthesized through a one-pot catalytic ring-opening metathesis polymerization (ROMP) and hydrogenation sequence. The crystallinity of these materials can be adjusted by varying the BSE concentration or the steric bulk of the Si-substituents, providing handles to control thermomechanical properties. Two methods for chemical recycling of HDPE mimics are introduced, including a circular approach that leverages acid-catalyzed Si−O bond exchange with 1-propanol. Additionally, despite the fact that the starting HDPE mimics were synthesized by chain-growth polymerization (ROMP), we show that it is possible to recover the molar mass and dispersity of recycled HDPE products using step-growth Si−O bond formation or exchange, generating high molecular weight recycled HDPE products with mechanical properties similar to commercial HDPE.
Metal Identity Effects in the Fracture Behavior of Coordinatively Crosslinked Elastomers
P. N. Johnson, Y. Yao, X. Huang, I. Kevlishvili, S. Schrettl, C. Weder, H. J. Kulik, and S. L. Craig
October 20, 2023

Polymers comprising polybutadiene backbones with 2,6-bis(1′-methyl-benzimidazolyl)pyridine (MeBip) sidechains were crosslinked by complexation with two different metal salts, either with copper(II) trifluoromethanosulfonate or with iron(II) trifluoromethanosulfonate. Dynamic mechanical analysis (DMA) and small-angle X-ray scattering (SAXS) data indicate that the crosslinking density and topology of the two materials are the same. The material crosslinked with copper ions, however, exhibits a higher extensibility and fracture energy than the polymer crosslinked with iron. These differences are attributed to differing mechanochemical responses of the metal complexes to applied stress. Computational results further indicate that the copper complexes are more labile, both in the stress-free state as well as upon application of force, and that the “open” complex in which only one MeBip ligand coordinates copper binds fewer counter-ions than the iron-coordinated analog. Both these factors enable easier re-binding of a second MeBip ligand. The computations further suggest that mechanochemically coupled spin-crossover behavior must be considered to fully understand the response of these metal-ligand complexes to mechanical stimuli. The data presented here furthers the facile manipulation of a material's strain response via metal species modulation, and the results offer a way to understand the relationship between bulk and molecular strain response.
Structure, Dynamics, and Rheology of Vitrimers
J. Xia, J. A. Kalow, and M. Olvera de la Cruz
September 29, 2023

Vitrimers are associative covalent adaptable networks that undergo reversible bond-exchange reactions while maintaining a fixed cross-linking density with changing temperature. To date, experimental studies that rely on macroscopic rheology have not been able to reveal topological changes and microscopic dynamics in these materials. Here, coarse-grained molecular dynamics simulations combined with a Monte Carlo method are implemented to investigate the topological structural changes, microscopic dynamics, and linear rheology of unentangled side-chain-linked vitrimers in conjunction with the sticky Rouse model (SRM). We find that there is a minor variation in the topological structure with temperature. The dynamic heterogeneities of the bond-exchange behavior and the system dynamics increase remarkably when approaching the topological freezing transition temperature Tv. Quantitative agreement between the simulation results and the SRM predictions is observed for the stress relaxation, elastic and loss moduli, and the relative mean-squared displacement, especially at the intermediate- and long-time or low-frequency regimes, where the time–temperature superposition principle is satisfied. We obtain a scaling collapse curve for the dynamic bond relaxation time, the zero-shear viscosity, and the horizontal shift factors without introducing any parameters, suggesting that the microscopic and macroscopic dynamics exhibit a similar relaxation behavior even in the presence of loop defects. Moreover, these results are in good agreement with those predicted by the SRM, indicating that the linear rheology of unentangled vitrimers with a fast bond-exchange rate can be analyzed via a single-chain approach based on the SRM.
Polymer Networks with Cubic, Mixed Pd(II) and Pt(II) M6L12 Metal–Organic Cage Junctions: Synthesis and Stress Relaxation Behavior
J. Zhao, E. Bobylev, D. Lundberg, N. Oldenhuis, H. Wang, I. Kevlishvili, S. L. Craig, H. J. Kulik, X. Li, and J. A. Johnson
September 29, 2023

Metal–organic cages/polyhedra (MOCs) are versatile building blocks for advanced polymer networks with properties that synergistically blend those of traditional polymers and crystalline frameworks. Nevertheless, constructing polyMOCs from very stable Pt(II)-based MOCs or mixtures of metal ions such as Pd(II) and Pt(II) has not, to our knowledge, been demonstrated, nor has exploration of how the dynamics of metal–ligand exchange at the MOC level may impact bulk polyMOC energy dissipation. Here, we introduce a new class of polymer metal–organic cage (polyMOC) gels featuring polyethylene glycol (PEG) strands of varied length cross-linked through bis-pyridyl-carbazole-based M6L12 cubes, where M is Pd(II), Pt(II), or mixtures thereof. We show that, while polyMOCs with varied Pd(II) content have similar network structures, their average stress–relaxation rates are tunable over 3 orders of magnitude due to differences in Pd(II)- and Pt(II)-ligand exchange rates at the M6L12 junction level. Moreover, mixed-metal polyMOCs display relaxation times indicative of intrajunction cooperative interactions, which stands in contrast to previous materials based on point metal junctions. Altogether, this work (1) introduces a novel MOC architecture for polyMOC design, (2) shows that polyMOCs can be prepared from mixtures of Pd(II)/Pt(II), and (3) demonstrates that polyMOCs display unique relaxation behavior due to their multivalent junctions, offering a strategy for controlling polyMOC properties independently of their polymer components.
Systematic Investigation of Silicon Substitution on Single Macromolecule Mechanics
K. E. Wentz; Y. Yao, I. Kevlishvili, T. B. Kouznetsova, B. A. Mediavilla, H. J. Kulik, S. L. Craig, and R. S. Klausen
August 18, 2023

Four unsaturated poly(carbooligosilane)s (P1–P4) were prepared via acyclic diene metathesis polycondensation of new oligosilane diene monomers (1–4). These novel polymers with varying main-chain Si incorporation have high trans internal olefin stereochemistry (ca. 80%) and molecular weights (9500–21,700 g mol–1). Postpolymerization epoxidation converted all alkene moieties to epoxides and rendered the polymers (P5–P8) more electrophilic, which allowed for single-molecule force spectroscopy studies via a modified atomic force microscope setup with a silicon tip and cantilever. The single-chain elasticity of the polycarbooligosilanes decreased with increasing numbers of Si–Si bonds, a finding reproduced by quantum chemical calculations.
Thiol-Triggered Deconstruction of Bifunctional Silyl Ether Terpolymers via an SNAr-Triggered Cascade
C. M. Brown, K. E. L. Husted, Y. Wang, L. J. Kilgallon, P. Shieh, H. Zafar, D. J. Lundberg, and J. A. Johnson
August 2, 2023

While Si-containing polymers can often be deconstructed using chemical triggers such as fluoride, acids, and bases, they are resistant to cleavage by mild reagents such as biological nucleophiles, thus limiting their end-of-life options and potential environmental degradability. Here, using ring-opening metathesis polymerization, we synthesize terpolymers of (1) a “functional” monomer (e.g., a polyethylene glycol macromonomer or dicyclopentadiene); (2) a monomer containing an electrophilic pentafluorophenyl (PFP) substituent; and (3) a cleavable monomer based on a bifunctional silyl ether Image ID:d3sc02868b-t1.gif. Exposing these polymers to thiols under basic conditions triggers a cascade of nucleophilic aromatic substitution (SNAr) at the PFP groups, which liberates fluoride ions, followed by cleavage of the backbone Si–O bonds, inducing polymer backbone deconstruction. This method is shown to be effective for deconstruction of polyethylene glycol (PEG) based graft terpolymers in organic or aqueous conditions as well as polydicyclopentadiene (pDCPD) thermosets, significantly expanding upon the versatility of bifunctional silyl ether based functional polymers.
Chemical Compatibilization, Macro-, and Microphase Separation of Heteroassociative Polymers
S. P. O. Danielsen
July 31, 2023

A mean-field equilibrium theory for reversible network formation due to heterotypic pairwise interactions in mixtures of associative polymers is extended via a weak inhomogeneity expansion to account for spatial fluctuations due to chemical incompatibility. We consider solutions and blends of polymers of types A and B with many associating groups per chain, and consider only A–B association between these groups. The structural correlations of the reversibly bonded polymers are accounted for by considering the Gaussian 4-arm star-like chain conformations between cross-links, which is analogous to an affine-network assumption.
Phase Separation and Gelation in Solutions and Blends of Heteroassociative Polymers
S. P. O. Danielsen, A. N. Semenov, and M. Rubinstein
July 10, 2023

An equilibrium statistical mechanical theory for the formation of reversible networks in two-component solutions of associative polymers is presented to account for the phase behavior due to hydrogen-bonding, metal–ligand, electrostatic, or other pairwise heterotypic associative interactions. We derive explicit analytical expressions for the binding statistics, gelation condition, and free energy, in which we consider polymers of types A and B with many associating groups per chain and consider only A–B association between the groups. The free energy is approximated at the mean-field level, considering overlapping polymer chains with an ideal gas of “stickers” capable of intermolecular association.
How a Chain Can Be Extended While Its Bonds Are Compressed
L. Sapir, J. Brock, D. Chen, Q. Liao, S. Panyukov, and M. Rubinstein
June 26, 2023

Extending polymer chains results in a positive chain tension, fch, primarily due to conformational restrictions. At the level of individual bonds, however, tension, fb, is either negative or positive and depends on both chain tension and bulk pressure. Typically, the chain and bond tension are assumed to be directly related. In specific systems, however, this dependence may not be intuitive, whereby fch increases while fb decreases; i.e., the entire chain is extended while bonds are compressed. Specifically, increasing the grafting density of a polymer brush results in chain extension along the direction perpendicular to the grafting surface while the underlying bonds are compressed. Similarly, upon compression of polymer networks, the extension of chains oriented in the “free” direction increases while their bonds are getting more compressed. We demonstrate this phenomenon in molecular dynamics simulations and explain it by the fact that the pressure contribution to fb is dominant over a wide range of network deformations and brush grafting densities.
Facile Mechanochemical Cycloreversion of Polymer Cross-linkers Enhances Tear Resistance
S. Wang, Y. Hu, T. B. Kouznetsova, L. Sapir, D. Chen, A. Herzog-Arbeitman, J. A. Johnson, M. Rubinstein, and S. L. Craig
June 22, 2023

The mechanical properties of covalent polymer networks often arise from the permanent end-linking or cross-linking of polymer strands, and molecular linkers that break more easily would likely produce materials that require less energy to tear. We report that cyclobutane-based mechanophore cross-linkers that break through force-triggered cycloreversion lead to networks that are up to nine times as tough as conventional analogs. The response is attributed to a combination of long, strong primary polymer strands and cross-linker scission forces that are approximately fivefold smaller than control cross-linkers at the same timescales. The enhanced toughness comes without the hysteresis associated with noncovalent cross-linking, and it is observed in two different acrylate elastomers, in fatigue as well as constant displacement rate tension, and in a gel as well as elastomers.
Tailoring Dynamic Hydrogels by Controlling Associative Exchange Rates
V. Zhang, J. V. Accardo, I. Kevlishvili, E. F. Woods, S. J. Chapman, C. T. Eckdahl, C. L. Stern, H. J. Kulik, and J. A. Kalow
June 7, 2023

We develop a suite of hydrogel cross-links that employs an associative exchange mechanism, allowing stress relaxation to be tuned independently from stiffness. We present a structure-reactivity-property relationship that enables reactivity and stress relaxation timescales to be estimated in silico for new structures. This work, therefore, opens new avenues to design and control associative dynamic hydrogels with targeted properties, overcoming the limitations of previous systems that have largely relied on dissociative exchange mechanisms.
Linear and Multivalent PEGylation of Tobacco Mosaic Virus and the Effects on its Biological Properties
R. M. Caballero, I. González-Gamboa, S. L. Craig, and N. F. Steinmetz
June 3, 2023

We investigate polyethylene glycol (PEG) coatings on tobacco mosaic virus (TMV), which was used as a model nanocarrier system, to evaluate the effects of linear and multivalent PEG coatings at varying chain lengths on serum protein adsorption, antibody recognition, and macrophage uptake.
Synthesis and Ring-Opening Metathesis Polymerization of a Strained trans-Silacycloheptene and Single-Molecule Mechanics of Its Polymer
H. Wakefield IV, I. Kevlishvili, K. E. Wentz, Y. Yao, T. B. Kouznetsova, S. J. Melvin, E. G. Amrosious, A. Herzog-Arbeitman, M. A. Siegler, J. A. Johnson, S. L. Craig, H. J. Kulik, and R. S. Klausen
April 5, 2023

The cis- and trans-isomers of a silacycloheptene were selectively synthesized by the alkylation of a silyl dianion, a novel approach to strained cycloalkenes. The trans-silacycloheptene (trans-SiCH) was significantly more strained than the cis isomer, as predicted by quantum chemical calculations and confirmed by crystallographic signatures of a twisted alkene. Each isomer exhibited distinct reactivity toward ring-opening metathesis polymerization (ROMP), where only trans-SiCH afforded high-molar-mass polymer under enthalpy-driven ROMP. Hypothesizing that the introduction of silicon might result in increased molecular compliance at large extensions, we compared poly(trans-SiCH) to organic polymers by single-molecule force spectroscopy (SMFS). Force-extension curves from SMFS showed that poly(trans-SiCH) is more easily overstretched than two carbon-based analogues, polycyclooctene and polybutadiene, with stretching constants that agree well with the results of computational simulations.
Effect of Strand Molecular Length on Mechanochemical Transduction on Elastomers Probed With Uniform Force Sensors
T. Ouchi, W. Wang, B. Silverstein, J. A. Johnson, and S. L. Craig
March 15, 2023

Here we address the question how the deformation and tension experienced by a strand is influenced by strand length through the use of mechanophore force probes with discrete molecular weights.
A Polyelectrolyte Handle for Single-Molecule Force Spectroscopy
J. Wang, T. B. Kouznetsova, J. Xia, F. Jiménez Ángeles, M. Olvera de la Cruz, and S. L. Craig
March 9, 2023

Here we report a polyelectrolyte handle for single-molecule force spectroscopy that offers a combination of high (several hundred pN) attachment forces, good (~4%) success in obtaining a high-force (>200 pN) attachment, a non-fouling detachment process that allows for repetition, and specific attachment locations along the polymer analyte.
Contribution of Unbroken Strands to the Fracture of Polymer Networks
S. Wang, S. Panyukov, S. L. Craig, and M. Rubinstein
March 7, 2023

We present a modified Lake–Thomas theory that accounts for the molecular details of network connectivity upon crack propagation in polymer networks.
Elasticity of Slide-Ring Gels
D. Chen, S. Panyukov, L. Sapir, and M. Rubinstein
February 24, 2023

We develop a single-chain model to account for the redistribution of monomers between network strands of a primary chain.
Conformation of Network Strands in Polymer Gels
H. K. Beech, J. A. Johnson, and B. D. Olsen
February 20, 2023

Small angle neutron scattering was used to measure single chain radii of gyration of end-linked polymer gels before and after cross-linking to calculate the prestrain, which is the ratio of the average chain size in a cross-linked network to that of a free chain in solution...
Remolding and Deconstruction of Industrial Thermosets via Carboxylic Acid-Catalyzed Bifunctional Silyl Ether Exchange
K. E. L. Husted, C. M. Brown, P. Shieh, I. Kevlishvili, S. L. Kristufek, H. Zafar, J. V. Accardo, J. C. Cooper, R. S. Klausen, H. J. Kulik, J. S. Moore, N. R. Sottos, J. A. Kalow, and J. A. Johnson
January 13, 2023

Here, we show that bifunctional silyl ether, i.e., R′O–SiR2–OR′′, (BSE)-based comonomers generate covalent adaptable network analogues of the industrial thermoset polydicyclopentadiene (pDCPD) through a novel BSE exchange process facilitated by the low-cost food-safe catalyst octanoic acid.
Covalent Mechanochemistry and Contemporary Polymer Network Chemistry: A Marriage in the Making
E. M. Lloyd, J. R. Vakil, Y. Yao, N. R. Sottos, and S. L. Craig
January 4, 2023

Over the past 20 years, the field of polymer mechanochemistry has amassed a toolbox of mechanophores that translate mechanical energy into a variety of functional responses ranging from color change...
Triplet Fusion Upconversion for Photocuring 3D Printed Particle-Reinforced Composite Networks
J. Wong, S. Wie, R. Meir, N. Sadaba, N. A. Ballinger, E. K. Harmon, X. Gao, G. Altin-Yavuzarslan, L. D. Pozzo, L. M. Campos, and A. Nelson
January 3, 2023

High energy photons (λ < 400 nm) are frequently used to initiate free radical polymerizations to form polymer networks, but are only effective for transparent objects. This phenomenon poses a major challenge...
Structure–Reactivity–Property Relationships in Covalent Adaptable Networks
V. Zhang, B. Kang , J. V. Accardo, and J. A. Kalow
November 29, 2022

In this Perspective, we analyze structure–reactivity–property relationships for several classes of CANs, illustrating both general design principles and the predictive potential of linear free energy relationships (LFERs) applied to CANs. We discuss opportunities in the field to develop quantitative structure–reactivity–property relationships and open challenges.
Efficient Manufacture, Deconstruction, and Upcycling of High-Performance Thermosets and Composites
E. M. Lloyd, J. C. Cooper, P. Shieh, D. G. Ivanoff, N. A. Parikh, E. B. Mejia, K. E. L. Husted, L. C. Costa, N. R. Sottos, J. A. Johnson, and J. S. Moore
November 16, 2022

Thermoset polymers and fiber-reinforced polymer composites possess the chemical, physical, and mechanical properties necessary for energy-efficient vehicles and structures, but their energy-inefficient...
Highly Efficient Bromine Capture and Storage Using N-Containing Porous Organic Cages
S. Lee, I. Kevlishvili, H. J. Kulik, H.-T. Kim, Y. G. Chung, and D.-Y. Koh
November 1, 2022

Highly volatile and toxic bromine (Br2) molecules can be utilized safely in various chemical processes when coupled with efficient separation systems. Herein, we present two different N-containing porous organic cages (POCs), covalent cage 3-R (CC3-R) and formaldehyde tied-reduced covalent cage 3 (FT-RCC3), for vapor Br2 capture under ambient conditions. They show outstanding sorption capacities (11.02 mmol g−1 and 11.64 mmol g−1, respectively) compared with previously reported adsorbents. Reversibility of the Br2 sorption process has been elucidated experimentally and computationally by identifying bromine species adsorbed at POCs and calculating their binding energies. The strong charge-transfer interactions between adsorbed Br2 and abundant N atomic sites of the host cages led to the dominant formation of polybromide species (Br3− and Br5−). Further host–guest interaction between POCs and polybromides determined the reversibility of the Br2 sorption process—showing partially reversible (>70% recovery) behavior for CC3-R and irreversible (<10% recovery) behavior for FT-RCC3, both of which were affected by the chemical and structural nature of different POCs. DFT calculations further indicate that the formation of carbocationic species (Br3− and Br5−) and HBr is energetically favorable within the cage, which is in good agreement with the experimental results. This work demonstrates that strong host–guest interactions are essential for highly efficient Br2 capture and storage performance.
Relaxation Dynamics of Supramolecular Polymer Networks with Mixed Cross-Linkers
D. Xu, B. D. Olsen, and S. L. Craig
November 1, 2022

The linear rheological properties of supramolecular polymer networks formed by mixtures of two different bis-Pd(II) cross-linkers with poly(4-vinylpyridine) in dimethyl sulfoxide are examined. The changes...
Extending BigSMILES to Non-Covalent Bonds in Supramolecular Polymer Assemblies
W. Zou, A. M. Monterroza, Y. Yao, S. C. Millik, M. M. Cencer, N. J. Rebello, H. K. Beech, M. A. Morris, T-S. Lin, C. S. Castano, J. A. Kalow, S. L. Craig, A. Nelson, J. S. Moore, and B. D. Olsen
September 15, 2022
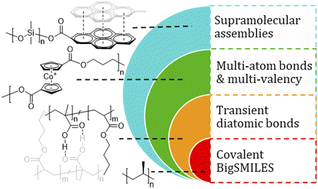
As a machine-recognizable representation of polymer connectivity, BigSMILES line notation extends SMILES from deterministic to stochastic structures. The same framework that allows BigSMILES...
Endohedrally Functionalized Metal–Organic Cage-Cross-Linked Polymer Gels as Modular Heterogeneous Catalysts
C. M. Brown, D. J. Lundberg, J. R. Lamb, I. Kevlishvili, D. Kleinschmidt, Y. S. Alfaraj, H. J. Kulik, M. F. Ottaviani, N. J. Oldenhuis, and J. A. Johnson
July 12, 2022

The immobilization of homogeneous catalysts onto supports to improve recyclability while maintaining catalytic efficiency is often a trial-and-error process limited by poor control of the local catalyst...
Scaling Theory of Swelling and Deswelling of Polymer Networks
T. Yamamoto, J. A. Campbell, S. Panyukov, and M. Rubinstein
April 20, 2022

We have developed a scaling theory of the elasticity of swollen and deswollen polymer networks. The elasticity of unentangled networks is primarily due to cross-links, and the elasticity of entangled...
Injectable Slow-Release Hydrogel Formulation of a Plant Virus-Based COVID-19 Vaccine Candidate
C .I. Nkanga, O. A. Ortega-Rivera, M. D. Shin, M. A. Moreno-Gonzalez, and N. F. Steinmetz
March 28, 2022

Cowpea mosaic virus (CPMV) is a potent immunogenic adjuvant and epitope display platform for the development of vaccines against cancers and infectious diseases, including coronavirus disease 2019...
Mechanically Triggered Carbon Monoxide Release with Turn-On Aggregation-Induced Emission
Y. Sun, W. J. Neary, Z. P. Burke, H. Qian, L. Zhu, and J. S. Moore
January 12, 2022

Polymers that release functional small molecules under mechanical stress potentially serve as next-generation materials for catalysis, sensing, and mechanochemical dynamic therapy. To further expand...
Toughening Hydrogels Through Force-triggered Chemical Reactions that Lengthen Polymer Strands
Z. Wang, X. Zheng, T. Ouchi, T. B. Kouznetsova, H. K. Beech, B. H. Bowser, S. Wang, J. A. Johnson, J. A. Kalow, B. D. Olsen, J. P. Gong, M. Rubinstein, and S. L. Craig
October 7, 2021

The utility and lifetime of materials made from polymer networks, including hydrogels, depend on their capacity to stretch and resist tearing. In gels and elastomers, those mechanical properties...
Molecular Characterization of Polymer Networks
S. P. O. Danielsen, H. K. Beech, B. M. El-Zaatari, X. Wang, D. J. Lundberg, G. Stoychev, L. Sapir, S. Wang, Z. Wang, T. Ouchi, P. N. Johnson, Y. Hu, S. L. Craig, J. A. Kalow, J. A. Johnson, B. D. Olsen, and M. Rubinstein
April 1, 2021

Polymer networks are complex systems consisting of molecular components. Whereas the properties of the individual components are typically well understood by most chemists, translating that chemical...
Single-event Spectroscopy and Unravelling Kinetics of Covalent Domains Based on Cyclobutane Mechanophores
B. H. Bowser, S. Wang, T. B. Kouznetsova, H. K. Beech, B. D. Olsen, M. Rubinstein, and S. L. Craig
March 30, 2021

Mechanochemical reactions that lead to an increase in polymer contour length have the potential to serve as covalent synthetic mimics of the mechanical unfolding of noncovalent “stored length” domains...
Mechanism Dictates Mechanics: A Molecular Substituent Effect in the Macroscopic Fracture of a Covalent Polymer Network
S. Wang, H. K. Beech, B. H. Bowser, T. B. Kouznetsova, B. D. Olsen, M. Rubinstein, and S. L. Craig
March 2, 2021

The fracture of rubbery polymer networks involves a series of molecular events, beginning with conformational changes along the polymer backbone and culminating with a chain scission reaction. Here...
PolyDAT: A Generic Schema for Polymer Characterization
T.-S. Lin, N. J. Rebello, H. K. Beech, Z. Wang, B. M. El-Zaatari, D. J. Lundberg, J. A. Johnson, J. A. Kalow, S. L. Craig, and B. D. Olsen
February 22, 2021

Polymers are stochastic materials that represent distributions of different molecules. In general, to quantify the distribution, polymer researchers rely on a series of chemical characterizations...
Cross-Linker Control of Vitrimer Flow
B. M. El-Zaatari, J. S. A. Ishibashi, and J. A. Kalow
April 8, 2020
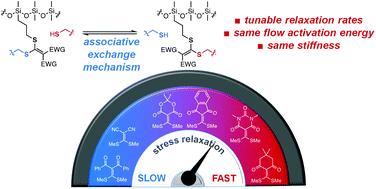
Vitrimers are a class of covalent adaptable networks (CANs) that undergo topology reconfiguration via associative exchange reactions, enabling reprocessing at elevated temperatures. Here, we show...
Photoswitchable Sol–Gel Transitions and Catalysis Mediated by Polymer Networks with Coumarin‐Decorated Cu24L24 Metal–Organic Cages as Junctions
N. J. Oldenhuis, P. Qin, S. Wang, H.-Z. Ye, E. Alt, A. Willard, T. V. Voorhis, S. L. Craig, and J. A. Johnson
November 19, 2019

Photoresponsive materials that change in response to light have been studied for a range of applications. These materials are often metastable during irradiation, returning to their pre-irradiated...
BigSMILES: A Structurally-Based Line Notation for Describing Macromolecules
T.-S. Lin, C. W. Coley, H. Mochigase, H. K. Beech, W. Wang, Z. Wang, E. Woods, S. L. Craig, J. A. Johnson, J. A. Kalow, K. F. Jensen, and B. D. Olsen
September 12, 2019

Having a compact yet robust structurally based identifier or representation system is a key enabling factor for efficient sharing and dissemination of research results within the chemistry community...
PolyMOF Nanoparticles: Dual Roles of a Multivalent polyMOF Ligand in Size Control and Surface Functionalization
Y. Gu, M. Huang, W. Zhang, M. A. Pearson, and J. A. Johnson
September 10, 2019

Metal–organic framework nanoparticles (MOF NPs) have emerged as an important class of materials that display significantly enhanced performance in many applications compared to bulk MOF materials...
Polymer Networks: From Plastics and Gels to Porous Frameworks
Y. Gu, J. Zhao, and J. A. Johnson
July 16, 2019

Polymer networks, which are materials composed of many smaller components—referred to as “junctions” and “strands”—connected together via covalent or non-covalent/supramolecular interactions...
Quantitative Adjustment to the Molecular Energy Parameter in the Lake-Thomas Theory of Polymer Fracture Energy
S. Wang, P. Sergey, M. Rubinstein, and S. L. Craig
March 21, 2019

We present a conceptual framework for adding molecular details of chain extension and force-coupled bond dissociation to the Lake–Thomas model of tear energy in rubbery crack propagation. Incorporating...
Follow Our Center's Research:
























